A DMRG-based Framework for Large-scale Quantum Many-body Calculations
Quantum chemical algorithms based on tensor factorizations are continuously expanding the scope of wave function-based molecular simulation methods. By leveraging very compact many-body wave function parametrizations, tensor network-based methods such as the density matrix renormalization group (DMRG) [1] can tame the computational cost of full configuration interaction (CI)-type calculations for strongly correlated molecular systems. While the DMRG algorithm is routinely applied to ground-state electronic structure problems, we present a versatile framework enabling its application to a broader range of many-body quantum problems [2,3]. We demonstrate the capabilities of our DMRG-based framework by introducing the n-mode vibrational DMRG method [4], which allows for an accurate treatment of the correlated nuclear problem for a reliable characterization of strongly anharmonic molecules described by complex potential energy surfaces. By expressing the vibrational CI wave function as a matrix product state, n-mode vDMRG can target systems with up to 30 fully coupled vibrational modes.
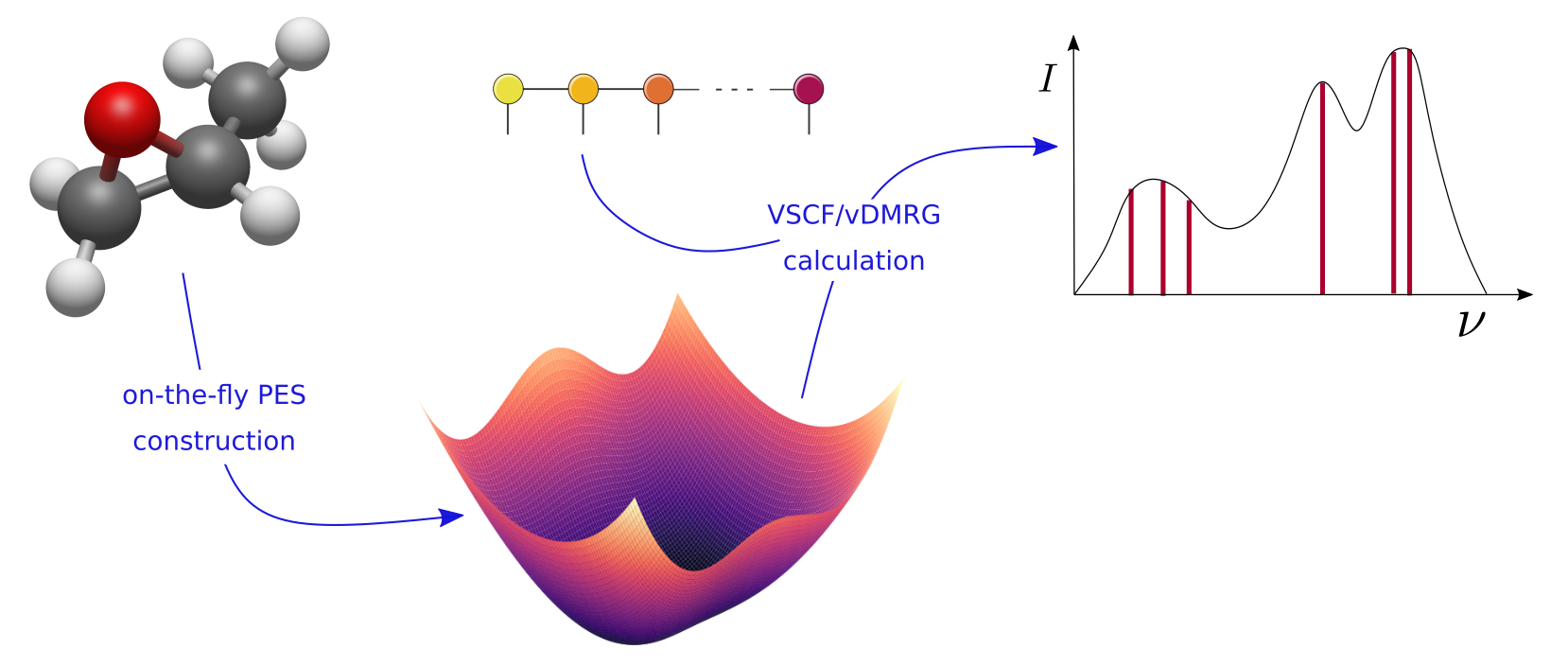
Figure 1: Illustration of the computational workflow of a n-mode vDMRG calculation
We further extend our framework to excited-states algorithms [5] enabling the large-scale calculation of both
low- and high-energy excitations. The combination of powerful tensor-based wave function representations
and efficient eigenstate targeting algorithms paves the route towards the accurate characterization of a
large variety of high-dimensional quantum systems.
[1] S. R. White, Phys. Rev. Lett., 1992, 69, 2863–2866.
[2] A. Baiardi, C. J. Stein, V. Barone, M. Reiher, J. Chem. Theory Comput., 2017, 13, 3764–3777.
[3] N. Glaser, A. Baiardi, M. Reiher. In Vibrational Dynamics of Molecules, World Scientific, 2022, 80-144.
[4] N. Glaser, A. Baiardi, M. Reiher, manuscript in preparation
[5] A. Baiardi, A. K. Kelemen, M. Reiher, J. Chem. Theory Comput., 2022, 18, 415–430.