Solvation Free Energies of Ion Dissociations in Dichloromethane: En Route to Accurate Computations
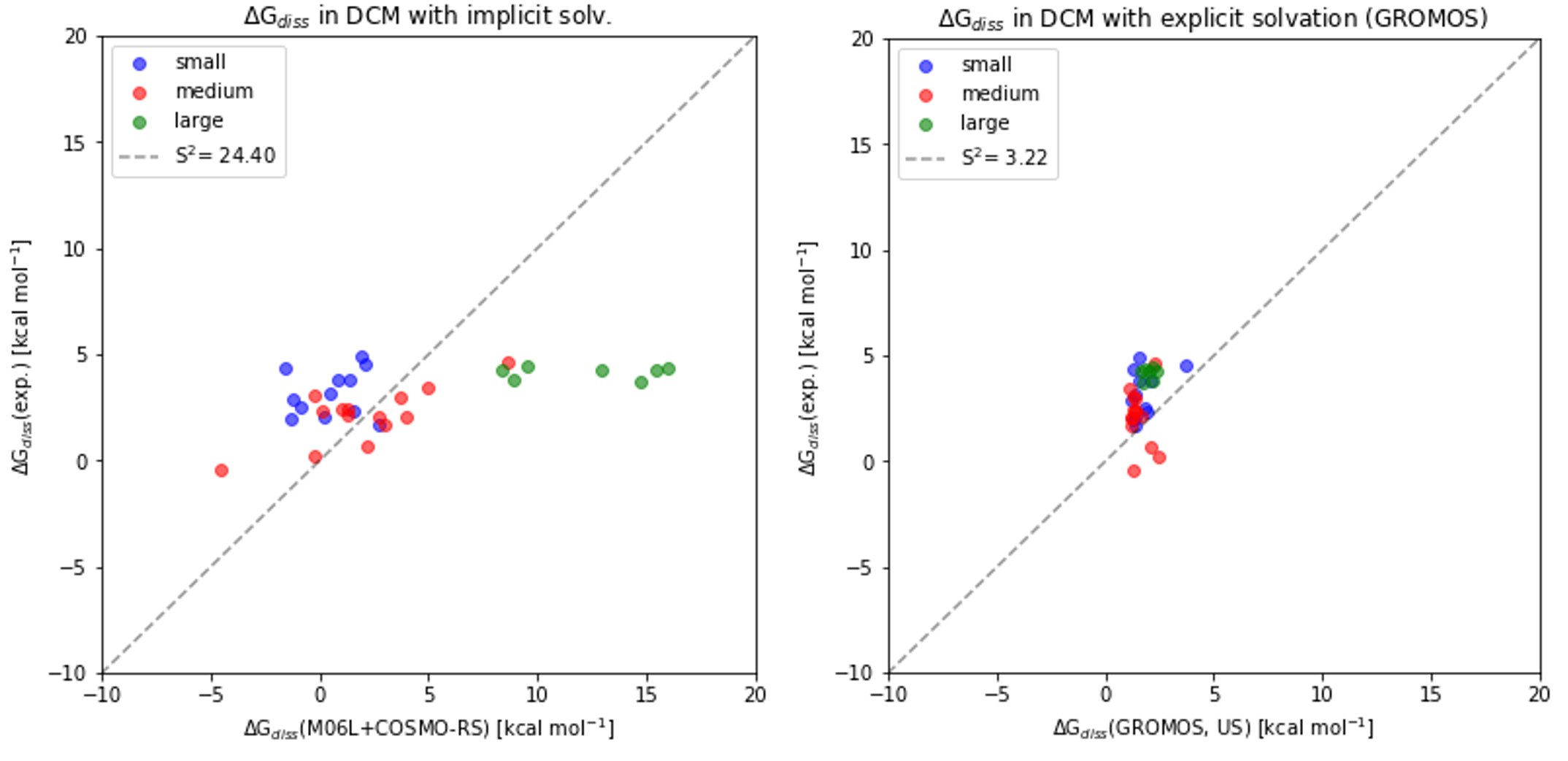
An extensive dataset of proton bound pyridine dimers was previously synthesized. An experimental and computational study of their dissociation, in the gas phase and in condensed phase, was previously reported [1][2]. Comparison of gas phase and condensed phase data gave considerable insights into solvation effects and the compensation of London dispersion interactions in condensed phase. State of the art DFT calculations and implicit solvent models failed spectacularly to reproduce free energies of dissociation in solvent, especially for larger quinoline dimers. We report a complementary computational study on the dissociation of proton-bound pyridine dimers in the gas phase and in dichloromethane (DCM). In an effort to determine the prerequisites to reproduce experimental trends and magnitudes of free energy of dissociation (∆Gdiss) in solvent, different contributions were investigated: The configurational and translational entropic contribution were estimated using xTB. Explicit solvation with classical mechanics was used as the most relevant method to treat not only the solute but also the solvent configurational entropy. This approach is a crude but fast computational method capable of predicting ∆Gdiss in DCM for this test system within a reasonable error margin. Despite the encouraging results in DCM, there are some limitations to the prediction of the free energy of solvation (∆Gsolv) which were unveiled and discussed extensively.
[1] R. Pollice, M. Bot, I. J. Kobylianskii, I. Shenderovich, P. Chen, J. Am. Chem. Soc., 2017, 139, 37, 13126–13140 [2] R. Pollice, F. Fleckenstein, I. Shenderovich, P. Chen, Angew. Chem. Int. Ed., 2019, 58, 40, 14281-14288